BAITS.IR module#
Import packages#
import os
import numpy as np
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
from scipy.stats import pearsonr
import BAITS
Import files#
Users could read the example data from the github, also could access the data through the link
bcr_loc_df = pd.read_csv('../data/bcr_sampled.tsv', sep='\t', index_col=0)
bcr_loc_df.head(n=2)
| sample | bcUmi | clone | aaClone | Vgene | Jgene | Cgene | cdr3nt | |
|---|---|---|---|---|---|---|---|---|
| 0 | P0516-LM | E14D5FEFEBE3D5C47 | TGTCAACAGAGTCACAGTATACCTACTTTT_CQQSHSIPTF_IGKV... | CQQSHSIPTF_IGKV1-39_IGKJ2_IGK | IGKV1-39 | IGKJ2 | IGK | TGTCAACAGAGTCACAGTATACCTACTTTT |
| 1 | P0516-LM | 8E153B79054792CA2 | TGTCAACAATATAGCACTGTCCCCCTCACTTTC_CQQYSTVPLTF_... | CQQYSTVPLTF_IGKV1-27_IGKJ4_IGK | IGKV1-27 | IGKJ4 | IGK | TGTCAACAATATAGCACTGTCCCCCTCACTTTC |
Define parameters#
sample_col = 'sample'
Umi_col = 'bcUmi'
clone_col = 'clone'
cdr3nt_col = 'cdr3nt'
Vgene_col = 'Vgene'
Jgene_col = 'Jgene'
Cgene_col = 'Cgene'
Quality Control#
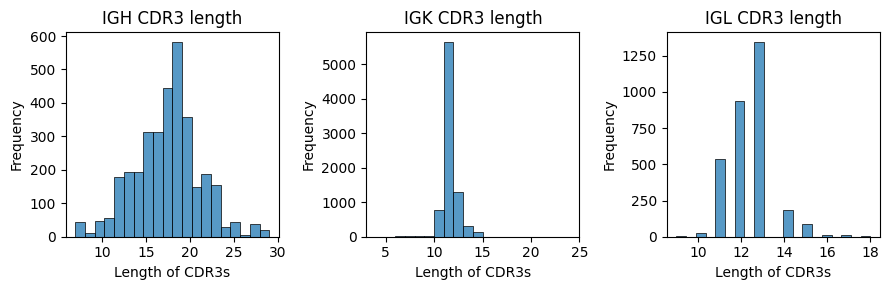
Calculate clonal CDR3 length#
Here, BAITS provides functions to calculate and visualize the length distribution of CDR3 region. The cauculated results will be returned.
bcr_loc_df = BAITS.VDJ.tl.calculate_cdr3_length(bcr_loc_df, sample_col, Cgene_col, cdr3nt_col, cdr3_type='nt', plot=True)
bcr_loc_df.head(n=2)
| sample | bcUmi | clone | aaClone | Vgene | Jgene | Cgene | cdr3nt | cdr3_length | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | P0516-LM | E14D5FEFEBE3D5C47 | TGTCAACAGAGTCACAGTATACCTACTTTT_CQQSHSIPTF_IGKV... | CQQSHSIPTF_IGKV1-39_IGKJ2_IGK | IGKV1-39 | IGKJ2 | IGK | TGTCAACAGAGTCACAGTATACCTACTTTT | 10 |
| 1 | P0516-LM | 8E153B79054792CA2 | TGTCAACAATATAGCACTGTCCCCCTCACTTTC_CQQYSTVPLTF_... | CQQYSTVPLTF_IGKV1-27_IGKJ4_IGK | IGKV1-27 | IGKJ4 | IGK | TGTCAACAATATAGCACTGTCCCCCTCACTTTC | 11 |
Calculate clonal richness#
BAITS provides function to calculate and visualize the clonal number per sample
bcr_loc_df = BAITS.VDJ.tl.stat_clone(bcr_loc_df, 'sample', Cgene_col, clone_col)
bcr_loc_df.head(n=2)
| sample | bcUmi | clone | aaClone | Vgene | Jgene | Cgene | cdr3nt | cdr3_length | clone_by_sample | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | P0516-LM | E14D5FEFEBE3D5C47 | TGTCAACAGAGTCACAGTATACCTACTTTT_CQQSHSIPTF_IGKV... | CQQSHSIPTF_IGKV1-39_IGKJ2_IGK | IGKV1-39 | IGKJ2 | IGK | TGTCAACAGAGTCACAGTATACCTACTTTT | 10 | 5216 |
| 1 | P0516-LM | 8E153B79054792CA2 | TGTCAACAATATAGCACTGTCCCCCTCACTTTC_CQQYSTVPLTF_... | CQQYSTVPLTF_IGKV1-27_IGKJ4_IGK | IGKV1-27 | IGKJ4 | IGK | TGTCAACAATATAGCACTGTCCCCCTCACTTTC | 11 | 5216 |
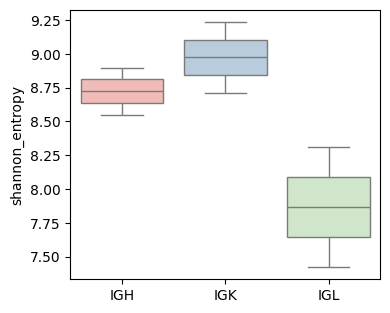
Calculate immune repertoire features#
Calculates the Shannon entropy of the CDR3 repertoire, leveraging UMI information to correct for sequencing errors and amplification bias.
groups = [sample_col, Cgene_col] # user-customizable based on experimental design
shannon_entropy_df = BAITS.VDJ.tl.compute_grouped_index(bcr_loc_df, sample_col, Cgene_col, clone_col, groups, count_basis = 'UMI', Umi_col='bcUmi', index='shannon_entropy')
Here, the Shannon entropy of the CDR3 repertoire is computed, stratified by both sample and C gene (chain type) groups.
shannon_entropy_df.head()
| sample | Cgene | shannon_entropy | |
|---|---|---|---|
| 0 | P0516-LM | IGH | 8.898825 |
| 1 | P0516-LM | IGK | 9.235398 |
| 2 | P0516-LM | IGL | 8.314985 |
| 3 | P1128-LM | IGH | 8.548061 |
| 4 | P1128-LM | IGK | 8.711763 |
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
BAITS.VDJ.pl._boxplot(shannon_entropy_df, 'Cgene', 'shannon_entropy', groupby='Cgene', palette='Pastel1', xlabel=None, ylabel='shannon_entropy', log=False, ax=ax )
plt.show()
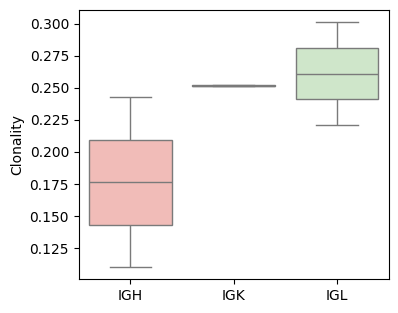
Calculates the Clonality of the CDR3 repertoire, leveraging UMI information to correct for sequencing errors and amplification bias.
Clonality quantifies the unevenness of clonal frequencies in the immune repertoire. Higher clonality suggests antigen-driven expansion and potential immune dominance
groups = [sample_col, Cgene_col]
shannon_entropy_df = BAITS.VDJ.tl.compute_grouped_index(bcr_loc_df, sample_col, Cgene_col, clone_col, groups, count_basis = 'UMI', Umi_col='bcUmi', index='Clonality')
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
BAITS.VDJ.pl._boxplot(shannon_entropy_df, 'Cgene', 'Clonality', groupby='Cgene', palette='Pastel1', xlabel=None, ylabel='Clonality', log=False, ax=ax )
plt.show()
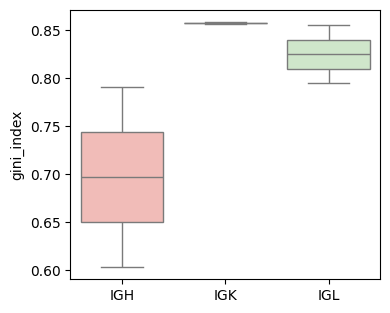
Gini_index (based on UMIs)
groups = [sample_col, Cgene_col]
gini_index_df = BAITS.VDJ.tl.compute_grouped_index(bcr_loc_df, sample_col, Cgene_col, clone_col, groups, count_basis = 'UMI', Umi_col='bcUmi', index='gini_index')
gini_index_df['tissue'] = gini_index_df['sample'].str.split('-').str[1]
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
BAITS.VDJ.pl._boxplot(gini_index_df, 'Cgene', 'gini_index', groupby='Cgene', palette='Pastel1', xlabel=None, ylabel='gini_index', log=False, ax=ax )
plt.show()
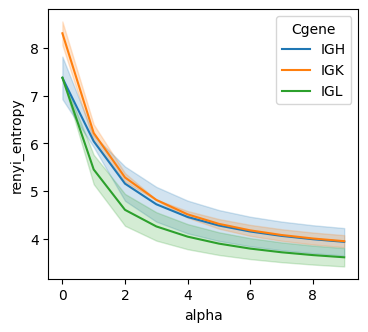
Renyi_entropy (based on UMIs)
Renyi entropy is a generalized diversity measure that captures different aspects of repertoire structure through an adjustable parameter alpha.
groups = [sample_col, Cgene_col]
renyi_entropy_df = BAITS.VDJ.tl.compute_grouped_index(bcr_loc_df, sample_col, Cgene_col, clone_col, groups, count_basis = 'UMI', Umi_col='bcUmi', index='renyi_entropy')
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
sns.lineplot(data=renyi_entropy_df, x="alpha", y="renyi_entropy", hue='Cgene', ax=ax )
plt.show()
Cluster BCR sequences to get BCR family#
We first focus on IGH BCRs (Immunoglobulin Heavy Chains) because they are the most important for analyzing BCR clusters and clonal families. By selecting only rows where the Cgene column equals ‘IGH’, we create a subset of the data that contains only IGH sequences, which will be used for downstream clustering and diversity analysis.
igh_bcr_loc_df = bcr_loc_df[bcr_loc_df['Cgene']=='IGH'].copy()
BCR sequences are first grouped by V gene, J gene, and CDR3 length. Within each group, sequences sharing ≥85% nucleotide identity (user-defined) are seen from the same BCR family.
igh_bcr_loc_df = BAITS.VDJ.tl.cluster_bcrs(igh_bcr_loc_df, threshold=0.85, sample_col='sample', Vgene_col='Vgene', Jgene_col='Jgene', cdr3nt_col='cdr3nt')
igh_bcr_loc_df.iloc[:,-5:].head()
| cdr3_length | clone_by_sample | CDR3_nt_length | BCR_familyID | Degree | |
|---|---|---|---|---|---|
| 0 | 13 | 1023 | 39 | IGHV1-45_IGHJ4_39_0 | 0 |
| 1 | 20 | 1023 | 60 | IGHV4-4_IGHJ5_60_0 | 0 |
| 2 | 14 | 1023 | 42 | IGHV4-39_IGHJ4_42_2 | 0 |
| 3 | 21 | 1023 | 63 | IGHV3-72_IGHJ4_63_0 | 1 |
| 4 | 18 | 1023 | 54 | IGHV1-69_IGHJ6_54_1 | 0 |
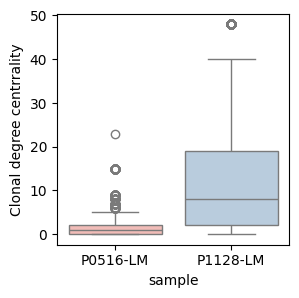
Degree centrality
Clonal degree centrality: Measure of the number of similar clones (with a hamming distance ≤ 1) connected to each unique V(D)J sequence within a clonal family. A high degree centrality value suggests that a particular V(D)J sequence serves as a progenitor clone, from which multiple related sequences have diverged via somatic hypermutation. This pattern is indicative of active antigen-driven diversification and affinity maturation. Conversely, a low degree centrality value indicates that the sequence is peripheral or isolated within the clonal network, with few connected variants. This may suggest limited subsequent diversification, potentially representing either a recently emerged clone or one that has not undergone significant antigen-driven expansion.
plt.subplots(1,1,figsize=(3, 3))
sns.boxplot(igh_bcr_loc_df, x='sample', y='Degree', palette='Pastel1')
plt.ylabel('Clonal degree centrrality')
plt.show()
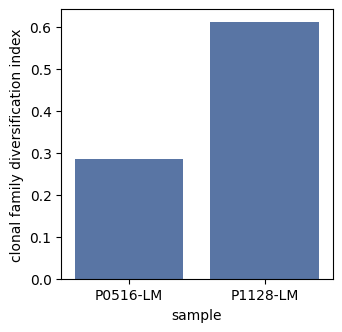
Calculate clonal family diversification index
Clonal family diversification: Measures the unevenness in the distribution of unique V(D)J sequences across clonal families using the Gini index. A high value indicates that certain clonal families contain numerous distinct V(D)J sequences – a pattern often associated with antigen-driven expansion and diversification.
Clonal_family_diver_idx = BAITS.VDJ.tl.Clonal_family_diversification(igh_bcr_loc_df, sample_col='sample', cluster_col='BCR_familyID', cdr3_col='cdr3nt')
Clonal_family_diver_idx.head()
| sample | Clonal_family_diver_index | |
|---|---|---|
| 0 | P0516-LM | 0.285553 |
| 1 | P1128-LM | 0.611375 |
plt.subplots(1,1,figsize=(3.5, 3.5))
sns.barplot(Clonal_family_diver_idx,x='sample', y='Clonal_family_diver_index', color='#4C72B0')
plt.ylabel('clonal family diversification index')
plt.show()