BAITS.st.module - Identify BLAs#
This tutorial shows an example of B lymphocyte aggregates (BLAs) identification. The dataset is composed of 2 human liver metastatic colorectal tumor samples profiled by Stereo-seq from originial paper.
Import packages and data#
import os
import BAITS
import anndata as ad
import numpy as np
import scanpy as sc
Users could access the data through the following link
adata = sc.read("./twosamples.h5ad")
adata
AnnData object with n_obs × n_vars = 968003 × 25091
obs: 'n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'celltype', 'sample'
var: 'mt'
obsm: 'spatial'
# Normalizing to median total counts
sc.pp.normalize_total(adata)
# Logarithmize the data
sc.pp.log1p(adata)
Identify B lymphocytes enriched regions#
To identify B cell-enriched regions in spatial transcriptomics data, we defined a custom gene signature. This signature was used to calculate an enrichment score for each spot, allowing for the spatial mapping of B cell locales.
signature = ["CD79A", "CD79B", "MS4A1", "CD79A", 'CD79B', "MZB1", "JCHAIN", "IGHA1", 'IGHG1', 'IGHG3']
Custom score name
score_name = 'Bcell_enrichment'
The user must provide the name of the column in adata.obs that identifies samples. Based on this parameter, BAITS performs integration by processing each sample separately, thereby reducing batch effects.
sample_name = 'sample'
Step1: Calculate the score#
sc.tl.score_genes(adata, gene_list=signature, score_name=score_name)
adata
AnnData object with n_obs × n_vars = 968003 × 25091
obs: 'n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'celltype', 'sample', 'Bcell_enrichment'
var: 'mt'
uns: 'log1p'
obsm: 'spatial'
Step2-1: Batch processing samples#
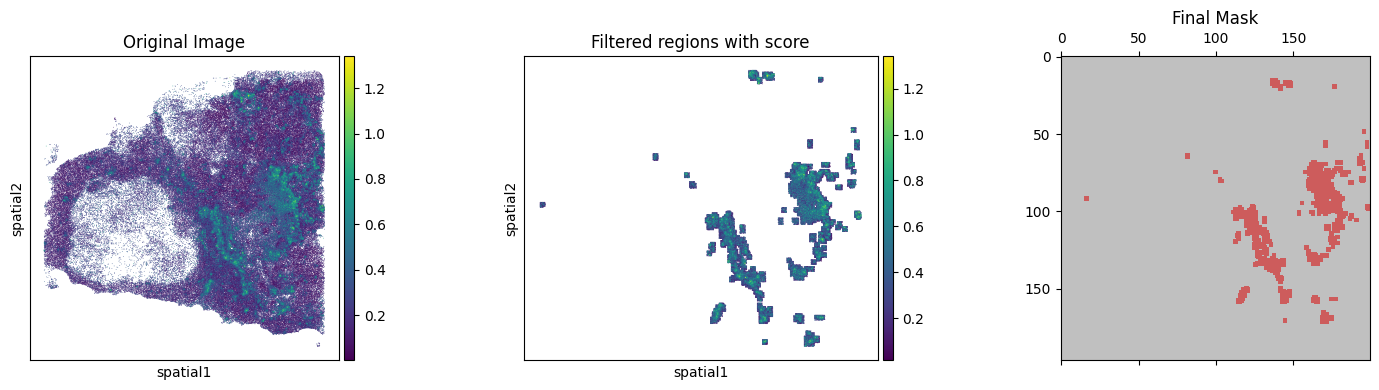
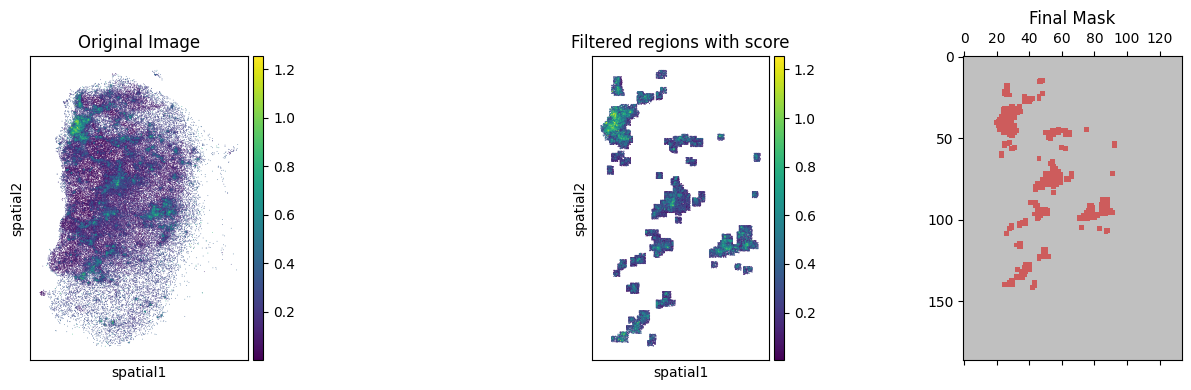
Using KDE algorithm to select the smoothed region with high B-cell score
adata = BAITS.st.tl.batch_process(
adata=adata, sample_name=sample_name, score_name=score_name,
processing_func = BAITS.st.tl.kde_filter,
verbose=True,
func_kwargs={ 'score_name':score_name, 'high_binSize': 100, 'default_thread_num': 36, 'clean_mask_size': (3,3)}
)
🔬 Start process sample: P1121_LM
🔎 Step 1 - Process data: 0.79 s
📊 Spatial cell number to evaluate: 39400
🏋️ Step 2 - KDE scoring: 335.07 s
📊 Using 90 th percentile threshold: 0.000
🔬 Step 3 - Obtain correct coords: 0.27 s
🎨 Plot results
✅ Sample: P1121_LM done!
🔬 Start process sample: P0516_LM
🔎 Step 1 - Process data: 0.09 s
📊 Spatial cell number to evaluate: 25058
🏋️ Step 2 - KDE scoring: 67.96 s
📊 Using 90 th percentile threshold: 0.000
🔬 Step 3 - Obtain correct coords: 0.10 s
🎨 Plot results
✅ Sample: P0516_LM done!
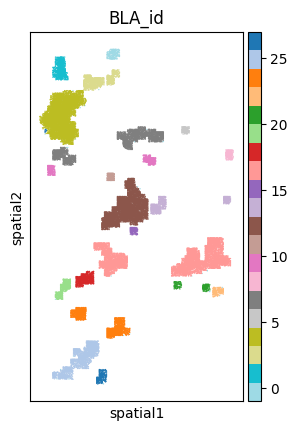
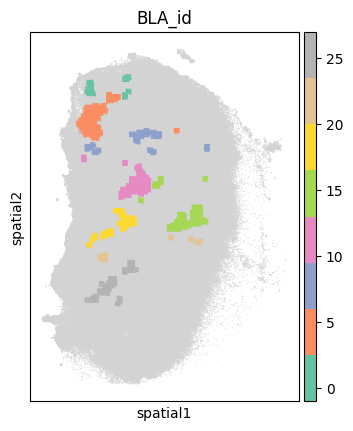
adata
AnnData object with n_obs × n_vars = 968003 × 25091
obs: 'n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'celltype', 'sample', 'Bcell_enrichment', 'filtered_coords'
obsm: 'spatial'
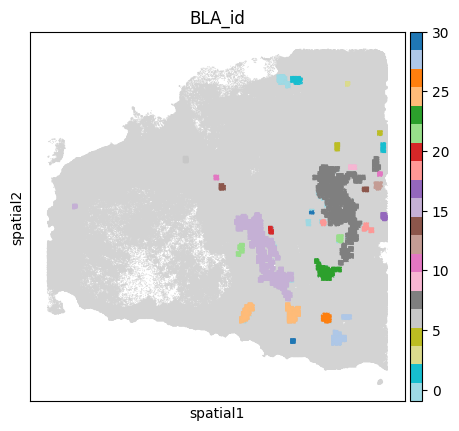
Using DBscan algorithm to cluster sinle BLA region
adata = BAITS.st.tl.batch_process(
adata=adata, sample_name=sample_name, score_name=score_name,
processing_func=BAITS.st.tl.dbscan_cluster,
verbose=True,
func_kwargs={ 'eps': 100, 'min_samples': 50 }
)
🔬 Start process sample: P1121_LM
✅ Sample: P1121_LM done!
🔬 Start process sample: P0516_LM
✅ Sample: P0516_LM done!
Step2-2: Processing samples one by one (Optional)#
To enable sample-specific fine-tuning, users can perform BAITS analysis on individual samples.
The analytical procedure is identical to the batch process described in Step 2-1; users may select their preferred configuration method.
# By using function **BAITS.st.tl.kde_filter**, we could filter cells with low B-cell-score
filtered_adata = []
for sample in ['P0516_LM']:
print('Sample:\t', sample)
tmp_ad = adata[adata.obs[sample_name]==sample].copy()
tmp_res = BAITS.st.tl.kde_filter(tmp_ad, score_name=score_name, plot=True, threshold_method='percentile', custom_threshold=90)
filtered_adata.append(tmp_res)
Sample: P0516_LM
🔎 Step 1 - Process data: 0.09 s
📊 Spatial cell number to evaluate: 25058
🏋️ Step 2 - KDE scoring: 66.45 s
📊 Using 90 th percentile threshold: 0.000
🔬 Step 3 - Obtain correct coords: 0.17 s
🎨 Plot results
adata = ad.concat(filtered_adata)
adata
AnnData object with n_obs × n_vars = 347386 × 25091
obs: 'n_genes_by_counts', 'total_counts', 'pct_counts_mt', 'celltype', 'sample', 'Bcell_enrichment', 'filtered_coords', 'BLA_id'
obsm: 'spatial'
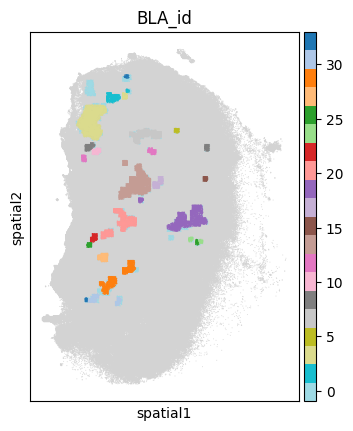
# **st.tl.dbscan_filter** employs the DBSCAN algorithm to identify spatially contiguous cellular domains into BLA.
res_adata = []
for sample in ['P0516_LM']:
print('Sample:\t', sample)
sample_ad = adata[adata.obs[sample_name]==sample].copy()
# select region with high B cell score
tmp_ad = sample_ad[sample_ad.obs['filtered_coords']].copy()
tmp_res = BAITS.st.tl.dbscan_cluster(tmp_ad)
sample_ad.obs['BLA_id'] = np.nan
sample_ad.obs.loc[tmp_res.obs.index, 'BLA_id'] = tmp_res.obs['BLA_id']
sc.pl.spatial(sample_ad, color='BLA_id', spot_size=50, cmap='Set2',
legend_fontsize='medium', legend_fontweight='normal', title='BLA_id', legend_loc='on data')
res_adata.append(sample_ad)
Sample: P0516_LM