BAITS.SR module#
import os
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
from scipy.stats import pearsonr
import matplotlib.pyplot as plt
import matplotlib.image as mpimg
import BAITS
Import files#
Users could read the example data from the github, also could access the data through the link
bcr_loc_df = pd.read_csv('./data/bcr_rep_loc_sampled.tsv', sep='\t',index_col=0)
bcr_loc_df.head()
| sample | bcUmi | clone | aaClone | Vgene | Jgene | Cgene | X | Y | spatial_cluster | tissue_region | InAggOrNot | Bcell_aggregate_label | BaggArea | celltype | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | P0516-LM | E14D5FEFEBE3D5C47 | TGTCAACAGAGTCACAGTATACCTACTTTT_CQQSHSIPTF_IGKV... | CQQSHSIPTF_IGKV1-39_IGKJ2_IGK | IGKV1-39 | IGKJ2 | IGK | 11873 | 3350 | C4: TLS-like | Peri-tumor | InAgg | P0516_LM-12 | 1789700.0 | B |
| 1 | P0516-LM | 8E153B79054792CA2 | TGTCAACAATATAGCACTGTCCCCCTCACTTTC_CQQYSTVPLTF_... | CQQYSTVPLTF_IGKV1-27_IGKJ4_IGK | IGKV1-27 | IGKJ4 | IGK | 7616 | 5287 | C4: TLS-like | Adjacent-tissue | InAgg | P0516_LM-12 | 1789700.0 | B |
| 2 | P0516-LM | CF7AAA1CE45FCED95 | TGTCAGCAATATTATGGTCTTCCATTCAGTTTG_CQQYYGLPFSL_... | CQQYYGLPFSL_IGKV4-1_IGKJ3_IGK | IGKV4-1 | IGKJ3 | IGK | 4077 | 2350 | C3: Plasma enriched | Adjacent-tissue | InAgg | P0516_LM-0 | 1098000.0 | Plasma |
| 3 | P0516-LM | 67DE18BD6C145817C | TGTCAGCAGTTTCACAACTGGCCCTACACATTT_CQQFHNWPYTF_... | CQQFHNWPYTF_IGKV3-15_IGKJ2_IGK | IGKV3-15 | IGKJ2 | IGK | 7390 | 5312 | C2: Macrophages enriched | Adjacent-tissue | InAgg | P0516_LM-12 | 1789700.0 | Hepatocytes |
| 4 | P0516-LM | 353E0FEC9BEE66A22 | TGTCAACAGTCCGCAAGTATTCCGTGGACGTTC_CQQSASIPWTF_... | CQQSASIPWTF_IGKV4-1_IGKJ1_IGK | IGKV4-1 | IGKJ1 | IGK | 4380 | 2441 | C3: Plasma enriched | Adjacent-tissue | InAgg | P0516_LM-0 | 1098000.0 | B |
Define parameters#
sample_col = 'sample'
Umi_col = 'bcUmi'
clone_col = 'clone'
cdr3nt_col = 'cdr3nt'
Vgene_col = 'Vgene'
Jgene_col = 'Jgene'
Cgene_col = 'Cgene'
loc_x_col = 'X'
loc_y_col='Y'
Quality Control#
Compute the clone number and UMIs of spatial location (at the bin1 level)
bcr_loc_df = BAITS.VDJ.tl.calculate_qc_clones( bcr_loc_df, sample_col, Cgene_col, clone_col, loc_x_col=loc_x_col, loc_y_col=loc_y_col, plot=True )
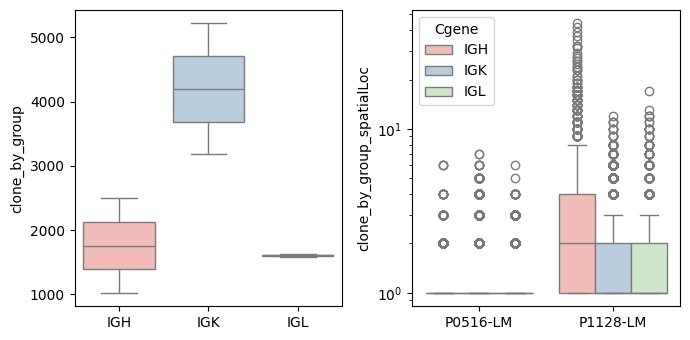
bcr_loc_df = BAITS.VDJ.tl.filter_clones_spatial(bcr_loc_df, 'clone_by_group_spatialLoc', min_clone_spatial=1)
bcr_loc_df = BAITS.VDJ.tl.calculate_qc_clones( bcr_loc_df, sample_col, Cgene_col, clone_col, plot=True )
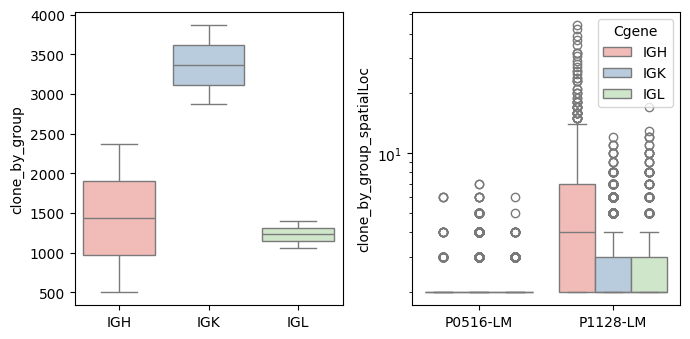
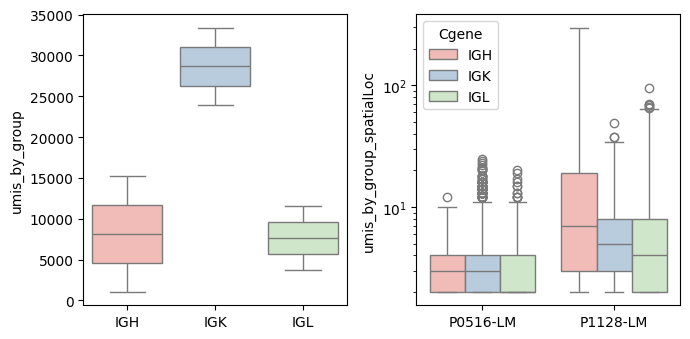
bcr_loc_df = BAITS.VDJ.tl.calculate_qc_umis( bcr_loc_df, sample_col, Cgene_col, clone_col, plot=False )
bcr_loc_df = BAITS.VDJ.tl.filter_umi_spatial(bcr_loc_df, 'umis_by_group_spatialLoc', min_umi_spatial=1)
bcr_loc_df = BAITS.VDJ.tl.calculate_qc_umis( bcr_loc_df, sample_col, Cgene_col, clone_col, plot=True )
Spatial repertoire feature#
For user-defined spatial regions of interest, such as B cell aggregates (BLAs) and other specialized spatial niches, this module calculates the full suite of BCR repertoire features introduced in the BAITS-IR module.
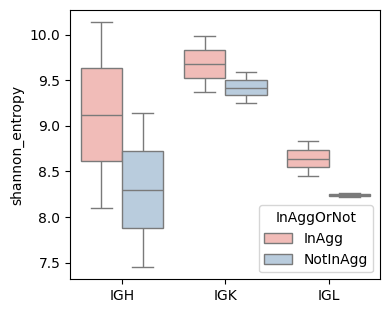
shannon_entropy (based on spatial location
groups = [sample_col, Cgene_col, 'InAggOrNot']
shannon_entropy_df = BAITS.VDJ.tl.compute_grouped_index(bcr_loc_df, sample_col, Cgene_col, clone_col, groups, loc_x_col=loc_x_col, loc_y_col=loc_y_col, index='shannon_entropy')
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
BAITS.VDJ.pl._boxplot(shannon_entropy_df, 'Cgene', 'shannon_entropy', groupby='InAggOrNot', palette='Pastel1', xlabel=None, ylabel='shannon_entropy', log=False, ax=ax )
plt.show()
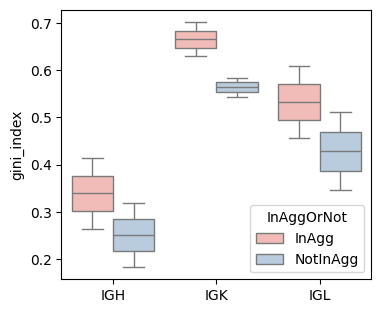
gini_index (based on spatial location
groups = [sample_col, Cgene_col, 'InAggOrNot']
gini_index_df = BAITS.VDJ.tl.compute_grouped_index(bcr_loc_df, sample_col, Cgene_col, clone_col, groups, loc_x_col=loc_x_col, loc_y_col=loc_y_col, index='gini_index')
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
BAITS.VDJ.pl._boxplot(gini_index_df, 'Cgene', 'gini_index', groupby='InAggOrNot', palette='Pastel1', xlabel=None, ylabel='gini_index', log=False, ax=ax )
plt.show()
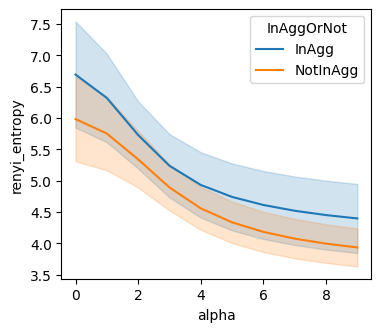
renyi_entropy (based on spatoal location
groups = [sample_col, Cgene_col, 'InAggOrNot']
renyi_entropy_df = BAITS.VDJ.tl.compute_grouped_index(bcr_loc_df, sample_col, Cgene_col, clone_col, groups, loc_x_col=loc_x_col, loc_y_col=loc_y_col, index='renyi_entropy')
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
sns.lineplot(data=renyi_entropy_df[renyi_entropy_df[Cgene_col]=='IGH'], x="alpha", y="renyi_entropy", hue='InAggOrNot', ax=ax )
plt.show()
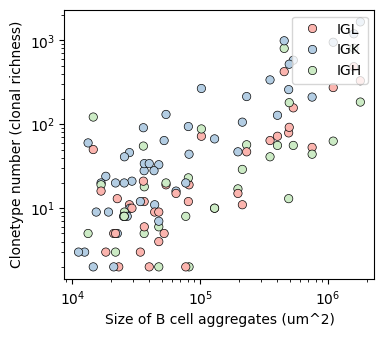
Corrlation between computed_index and the area of the B_aggregates
agg_clone_df = BAITS.VDJ.tl.calculate_qc_clones( bcr_loc_df, 'Bcell_aggregate_label', Cgene_col, clone_col, plot=False )
plot_df = agg_clone_df[['sample', 'Cgene', 'BaggArea','clone_by_group']].drop_duplicates().dropna()
fig, ax = plt.subplots(1,1,figsize=(4, 3.5))
BAITS.VDJ.pl._scatter_plot(plot_df, 'BaggArea', 'clone_by_group', groupby='Cgene', palette='Pastel1', x_log=True, y_log=True,
xlabel='Size of B cell aggregates (um^2)', ylabel='Clonetype number (clonal richness)', ax=ax )
plt.show()
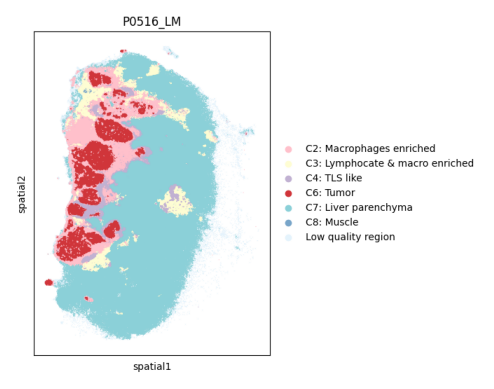
Clonal migration#
In the tutorial “st_spatial_cluster”, we define the niches within a region through spatial clustering, as illustrated below:
img = mpimg.imread('/home/zhaoyp/1.CRLM_XCR/BAITS/docs/data/P0516_LM_spatialCluster_umap.png')
plt.imshow(img)
plt.axis('off')
plt.show()
By the way, we also could visualize the spatial distribution of VDJ sequences, as illustrated below:
BAITS.VDJ.pl._plot_xcr( bcr_loc_df[(bcr_loc_df[sample_col]=='P0516-LM') & (bcr_loc_df[Cgene_col]=='IGH')], clone_col,loc_x_col, loc_y_col )
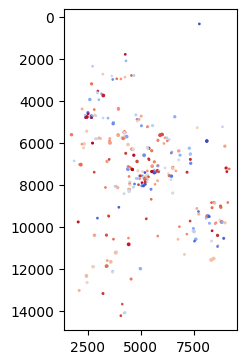
Based on spatial niches and VDJ distribution, we could use spatial migration index to quantify the BCR ‘migration’ patten.
migr_df = BAITS.VDJ.tl.compute_migraIdx(bcr_loc_df, sample_col='sample', clone_col='clone', chain_col='Cgene', cluster_col='spatial_cluster', x_col='X', y_col='Y')
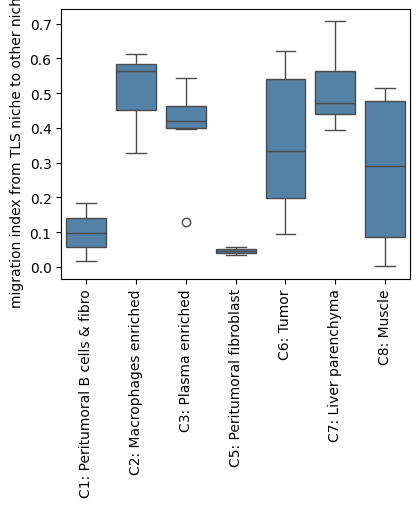
The clonal migration index from the TLS region to other niches is visualized here.
tls_to_otherNiche = migr_df[migr_df['cluster_source']=='C4: TLS-like'].sort_values('cluster_target').copy()
plt.subplots(1, 1, figsize=(4.5, 3.5))
sns.boxplot(data=tls_to_otherNiche, x='cluster_target', y='BCR_cross',color='steelblue')
plt.xticks(rotation=90)
plt.ylabel('migration index from TLS niche to other niches')
plt.xlabel('')
plt.show()
Clonal niche analysis#
To investigate the spatial niche surrounding the specific clone, we filtered the most expanded clones.
bcr_loc_df[['sample','aaClone']].value_counts().head(2)
sample aaClone
P0516-LM CQQYHDLPITF_IGKV1-33_IGKJ5_IGK 2132
CQQSYRTPRLLFTF_IGKV1-39_IGKJ3_IGK 1471
dtype: int64
Here, we investigate the spatial niche surrounding the most expanded clone, ‘CQQYHDLPITF_IGKV1-33_IGKJ5_IGK’, in sample ‘P0516-LM’.
data_out, niche_prop, niche_count = BAITS.VDJ.tl.calculate_clone_niche(
bcr_loc_df,
sample="P0516-LM",
target_clone="CQQYHDLPITF_IGKV1-33_IGKJ5_IGK",
radius=50, # um
scale=2 # In Stereo-seq, the length of each spot is 0.5 um
)
import matplotlib.pyplot as plt
import seaborn as sns
fig, ax = plt.subplots(1, 1, figsize=(4.5, 3.5))
sns.barplot(x=niche_prop.index, y=niche_prop.values, color='steelblue', ax=ax)
ax.set_ylabel('Surrounding cell type proportion')
ax.set_xlabel('')
plt.xticks(rotation=90)
for i, v in enumerate(niche_prop.values):
ax.text( i, v + 0.005, f"{v:.2f}", ha='center',va='bottom',fontsize=9)
plt.tight_layout()
plt.show()
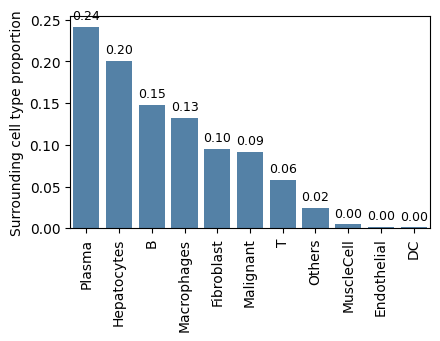
Here, we observe that the most abundant cell type surrounding the clone ‘CQQYHDLPITF_IGKV1-33_IGKJ5_IGK’ in sample ‘P0516-LM’ is ‘plasma’, followed by ‘hepatocytes’.